Case Study 2: Mouse hippocampus development¶
This tutorial shows how cellDancer analyzes RNA velocity. The process includes (1) estimating RNA velocity, (2) deriving cell fates on embedding space, (3) estimating pseudotime, and (4) displaying predicted kinetics rates (transcription, splicing, and degradation rates), spliced mRNA abundance, and unspliced mRNA abundance on embedding space.
Below is the case study for the mouse hippocampal dentate gyrus neurogenesis. We follow the preprocessing methods of La Manno et al. to filter genes and cells. 18,140 cells with 2,159 genes were selected.
Import packages¶
To run the notebook locally, Installation could be referred to install the environment and dependencies.
[1]:
# import packages
import os
import sys
import glob
import pandas as pd
import math
import matplotlib.pyplot as plt
import celldancer as cd
import celldancer.cdplt as cdplt
from celldancer.cdplt import colormap
Load data¶
The input data for cellDancer contains the preprocessed abundances of unspliced RNA and spliced RNA. The data of mouse hippocampal dentate gyrus neurogenesis can be ownloaded from DentateGyrus_cell_type_u_s.csv.zip. To load your own data, the dataframe should contain columns ‘gene_name’, ‘unsplice’, ‘splice’ ,‘cellID’ ,‘clusters’ ,‘embedding1’, and ‘embedding2.’ For a detailed description about the preprocessing and the data structure, refer to Data Preparation.
[2]:
cell_type_u_s_path="your_path/DentateGyrus_cell_type_u_s.csv"
cell_type_u_s=pd.read_csv(cell_type_u_s_path)
cell_type_u_s
[2]:
| gene_name | unsplice | splice | cellID | clusters | embedding1 | embedding2 | |
|---|---|---|---|---|---|---|---|
| 0 | Rgs20 | 0.069478 | 0.021971 | 10X83_2:AAACGGGGTCTCTTTAx | ImmGranule2 | 18.931086 | -1.862429 |
| 1 | Rgs20 | 0.085834 | 0.016256 | 10X83_2:AACCATGGTTCAACCAx | ImmGranule2 | 18.419891 | -1.282895 |
| 2 | Rgs20 | 0.068644 | 0.047774 | 10X83_2:AACACGTTCTGAAAGAx | CA2-3-4 | 2.369887 | 16.868419 |
| 3 | Rgs20 | 0.045387 | 0.018101 | 10X83_2:AAAGATGCATTGAGCTx | CA | -5.351040 | 10.676485 |
| 4 | Rgs20 | 0.040457 | 0.012846 | 10X83_2:AACCATGTCTACTTACx | CA1-Sub | -6.189126 | 11.754900 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 39164255 | Gpm6b | 0.876650 | 1.276089 | 10X84_3:TTTCCTCCACCATCCTx | ImmGranule1 | 10.812611 | -2.487668 |
| 39164256 | Gpm6b | 2.024897 | 5.152006 | 10X84_3:TTTGTCACATGAAGTAx | CA2-3-4 | 8.246204 | 23.482788 |
| 39164257 | Gpm6b | 1.848051 | 1.491445 | 10X84_3:TTTCCTCCACGGTAAGx | nIPC | -3.441272 | -4.917364 |
| 39164258 | Gpm6b | 0.696361 | 1.189091 | 10X84_3:TTTGTCAAGCGTCAAGx | ImmGranule2 | 16.394199 | -6.143549 |
| 39164259 | Gpm6b | 0.739348 | 1.239052 | 10X84_3:TTTCCTCGTGAAAGAGx | ImmGranule2 | 17.490857 | -4.130190 |
39164260 rows × 7 columns
Estimate RNA velocity for sample genes¶
We use cd.velocity() to estimate the velocity. Here, 30 genes in gene_list are estimated as an example. The predicted unspliced and spliced reads, alpha, beta, and gamma are added to the dataframe.
[3]:
gene_list=['Psd3', 'Dcx', 'Syt11', 'Ntrk2', 'Gnao1', 'Gria1', 'Dctn3', 'Map1b', 'Camk2a', 'Gpm6b', 'Sez6l', 'Evl', 'Astn1', 'Ank2', 'Klf7', 'Tbc1d16', 'Atp1a3', 'Stxbp6', 'Scn2a1', 'Lhx9', 'Slc4a4', 'Ppfia2', 'Kcnip1', 'Ptpro', 'Diaph3', 'Slc1a3', 'Cadm1', 'Mef2c', 'Sptbn1', 'Ncald']
loss_df, cellDancer_df=cd.velocity(cell_type_u_s,
gene_list=gene_list,
permutation_ratio=0.1,
norm_u_s=False,
norm_cell_distribution=False,
n_jobs=8)
cellDancer_df
Using /Users/shengyuli/Library/CloudStorage/OneDrive-HoustonMethodist/work/Velocity/bin/cellDancer_polish/analysis/CaseStudyNotebook/cellDancer_velocity_2022-06-27 16-37-44 as the output path.
Arranging genes for parallel job.
30 genes were arranged to 4 portions.
Velocity Estimation: 0%| | 0/4 [00:00<?, ?it/s]
Velocity Estimation: 25%|██▌ | 1/4 [00:37<01:53, 37.68s/it]
Velocity Estimation: 50%|█████ | 2/4 [01:04<01:03, 31.53s/it]
Velocity Estimation: 75%|███████▌ | 3/4 [01:31<00:29, 29.08s/it]
Velocity Estimation: 100%|██████████| 4/4 [01:52<00:00, 26.11s/it]
[3]:
| cellIndex | gene_name | unsplice | splice | unsplice_predict | splice_predict | alpha | beta | gamma | loss | cellID | clusters | embedding1 | embedding2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | Klf7 | 0.408467 | 1.294797 | 0.444935 | 1.475828 | 0.454905 | 0.935128 | 0.015373 | 0.077489 | 10X83_2:AAACGGGGTCTCTTTAx | ImmGranule2 | 18.931086 | -1.862429 |
| 1 | 1 | Klf7 | 0.379136 | 1.256870 | 0.411796 | 1.424216 | 0.419835 | 0.935061 | 0.015772 | 0.077489 | 10X83_2:AACCATGGTTCAACCAx | ImmGranule2 | 18.419891 | -1.282895 |
| 2 | 2 | Klf7 | 0.893599 | 3.395004 | 0.969404 | 3.832591 | 1.033540 | 0.986942 | 0.001990 | 0.077489 | 10X83_2:AACACGTTCTGAAAGAx | CA2-3-4 | 2.369887 | 16.868419 |
| 3 | 3 | Klf7 | 0.640505 | 2.739187 | 0.669036 | 3.047821 | 0.684626 | 0.979797 | 0.003759 | 0.077489 | 10X83_2:AAAGATGCATTGAGCTx | CA | -5.351040 | 10.676485 |
| 4 | 4 | Klf7 | 0.662303 | 2.433943 | 0.712970 | 2.749427 | 0.745024 | 0.971894 | 0.005226 | 0.077489 | 10X83_2:AACCATGTCTACTTACx | CA1-Sub | -6.189126 | 11.754900 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 544195 | 18135 | Kcnip1 | 0.018745 | 0.005679 | 0.243844 | 0.008779 | 0.463909 | 0.731405 | 1.322285 | 0.079982 | 10X84_3:TTTCCTCCACCATCCTx | ImmGranule1 | 10.812611 | -2.487668 |
| 544196 | 18136 | Kcnip1 | 0.148039 | 0.093618 | 0.380534 | 0.085685 | 0.572677 | 0.727422 | 1.319759 | 0.079982 | 10X84_3:TTTGTCACATGAAGTAx | CA2-3-4 | 8.246204 | 23.482788 |
| 544197 | 18137 | Kcnip1 | 0.080708 | 0.032079 | 0.312729 | 0.040230 | 0.522807 | 0.728119 | 1.323665 | 0.079982 | 10X84_3:TTTCCTCCACGGTAAGx | nIPC | -3.441272 | -4.917364 |
| 544198 | 18138 | Kcnip1 | 0.078976 | 0.033418 | 0.310343 | 0.040067 | 0.520257 | 0.728360 | 1.323348 | 0.079982 | 10X84_3:TTTGTCAAGCGTCAAGx | ImmGranule2 | 16.394199 | -6.143549 |
| 544199 | 18139 | Kcnip1 | 0.050686 | 0.019074 | 0.279029 | 0.024949 | 0.493671 | 0.729688 | 1.322998 | 0.079982 | 10X84_3:TTTCCTCGTGAAAGAGx | ImmGranule2 | 17.490857 | -4.130190 |
544200 rows × 14 columns
Visualize the phase portraits¶
We visualize the phase portrait of each gene with cdplt.scatter_gene().
[4]:
ncols=5
height=math.ceil(len(gene_list)/ncols)*4
fig = plt.figure(figsize=(20,height))
for i in range(len(gene_list)):
ax = fig.add_subplot(math.ceil(len(gene_list)/ncols), ncols, i+1)
cdplt.scatter_gene(
ax=ax,
x='splice',
y='unsplice',
cellDancer_df=cellDancer_df,
custom_xlim=None,
custom_ylim=None,
colors=colormap.colormap_neuro,
alpha=0.5,
s = 10,
velocity=True,
gene=gene_list[i])
ax.set_title(gene_list[i])
ax.axis('off')
plt.show()

Project the RNA velocity onto the embedding space¶
To project the prediction of RNA velocity to velocity onto the embedding space and to estimate pseudotime by using all genes, predicted result can be downloaded from DentateGyrus_cellDancer_estimation.csv.zip.
[5]:
# load the prediction result of all genes
cellDancer_df_file = 'your_path/DentateGyrus_cellDancer_estimation.csv'
cellDancer_df=pd.read_csv(cellDancer_df_file)
We calculate the projection of RNA velocity on the embedding with cd.compute_cell_velocity(). The projected direction on embedding space, i.e. columns ‘velocity1’ and ‘velocity2’ are added to the original dataframe. We use cdplt.scatter_cell() to display the predicted direction on embedding space.
[6]:
# compute cell velocity
cellDancer_df=cd.compute_cell_velocity(cellDancer_df=cellDancer_df)
# plot cell velocity
fig, ax = plt.subplots(figsize=(15,15))
im = cdplt.scatter_cell(ax,cellDancer_df,
colors=colormap.colormap_neuro,
alpha=0.3,
s=10,
velocity=True,
legend='on',
min_mass=2,
arrow_grid=(30,30))
ax.axis('off')
plt.show()

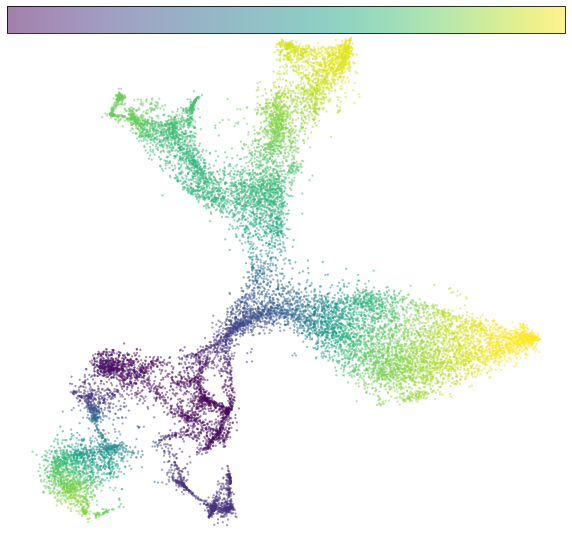
Estimate pseudotime¶
Based on the projection of RNA velocity on embedding space, we estimate the pseudotime with cd.pseudo_time().
[7]:
%%capture
# set parameters
dt = 0.001
t_total = {dt: 10000}
n_repeats = 10
# estimate pseudotime
cellDancer_df = cd.pseudo_time(cellDancer_df=cellDancer_df,
grid=(30, 30),
dt=dt,
t_total=t_total[dt],
n_repeats=n_repeats,
speed_up=(60,60),
n_paths = 5,
psrng_seeds_diffusion=[i for i in range(n_repeats)],
n_jobs=8)
[8]:
# plot pseudotime
fig, ax = plt.subplots(figsize=(10,10))
im=cdplt.scatter_cell(ax,cellDancer_df, colors='pseudotime', alpha=0.5, velocity=False)
ax.axis('off')
plt.show()
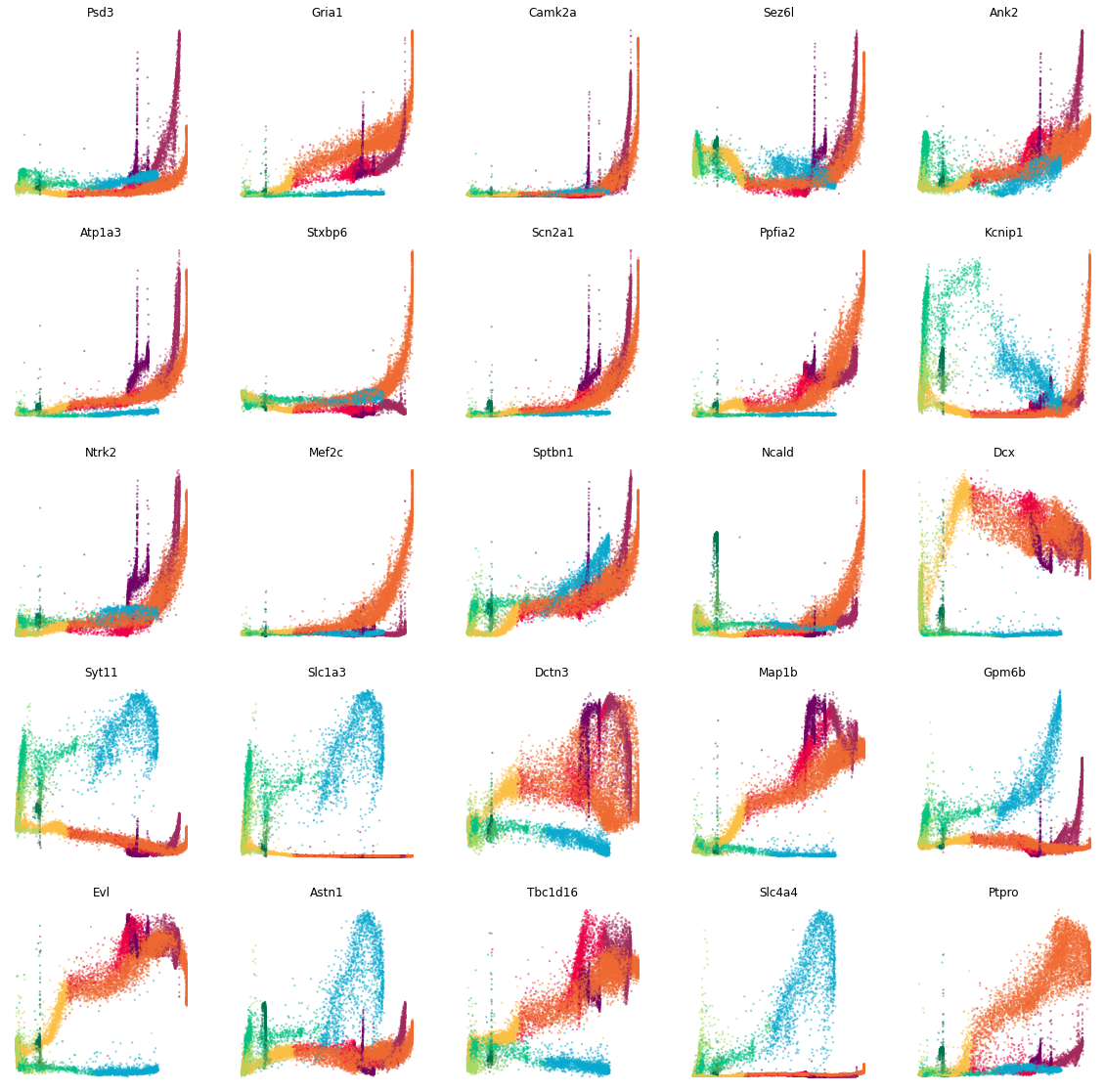
Display the abundance of spliced RNA along pseudotime¶
We visualize the spliced RNA abundance of some sample genes along pseudotime with cdplt.scatter_gene().
[9]:
gene_list=['Psd3', 'Gria1', 'Camk2a', 'Sez6l', 'Ank2', 'Atp1a3', 'Stxbp6', 'Scn2a1', 'Ppfia2', 'Kcnip1', 'Ntrk2', 'Mef2c', 'Sptbn1', 'Ncald','Dcx', 'Syt11','Slc1a3', 'Dctn3', 'Map1b', 'Gpm6b', 'Evl', 'Astn1', 'Tbc1d16','Slc4a4', 'Ptpro']
ncols=5
height=math.ceil(len(gene_list)/ncols)*4
fig = plt.figure(figsize=(20,height))
for i in range(len(gene_list)):
ax = fig.add_subplot(math.ceil(len(gene_list)/ncols), ncols, i+1)
cdplt.scatter_gene(
ax=ax,
x='pseudotime',
y='splice',
cellDancer_df=cellDancer_df,
custom_xlim=None,
custom_ylim=None,
colors=colormap.colormap_neuro,
alpha=0.5,
s = 5,
velocity=False,
gene=gene_list[i])
ax.set_title(gene_list[i])
ax.axis('off')
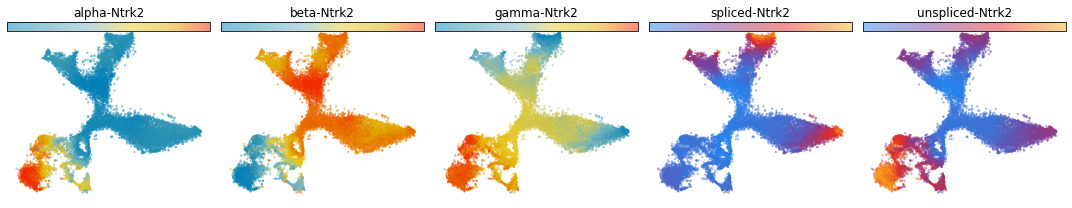
Visualize the reaction rates, the abundance of unspliced RNA, and the abundance of spliced RNA on embedding space¶
cellDancer provides other functions for visualization. we show the predicted kinetics rates (transcription, splicing, and degradation rates), spliced mRNA abundance, and unspliced mRNA abundance on embedding space with cdplt.scatter_cell().
[10]:
gene_samples=['Ntrk2','Psd3','Dcx']
for gene in gene_samples:
fig, ax = plt.subplots(ncols=5, figsize=(15,3))
cdplt.scatter_cell(ax[0],cellDancer_df, colors='alpha',
gene=gene, velocity=False)
cdplt.scatter_cell(ax[1],cellDancer_df, colors='beta',
gene=gene, velocity=False)
cdplt.scatter_cell(ax[2],cellDancer_df, colors='gamma',
gene=gene, velocity=False)
cdplt.scatter_cell(ax[3],cellDancer_df, colors='splice',
gene=gene, velocity=False)
cdplt.scatter_cell(ax[4],cellDancer_df, colors='unsplice',
gene=gene, velocity=False)
ax[0].axis('off')
ax[1].axis('off')
ax[2].axis('off')
ax[3].axis('off')
ax[4].axis('off')
ax[0].set_title('alpha-'+gene)
ax[1].set_title('beta-'+gene)
ax[2].set_title('gamma-'+gene)
ax[3].set_title('spliced-'+gene)
ax[4].set_title('unspliced-'+gene)
plt.tight_layout()
plt.show()